维生素B12全合成
百科内容来自于:

分子结构
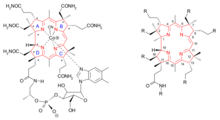
逆合成分析

合成路线
A环合成
A环的合成首先由间甲氧基苯胺(
1)和乙偶姻(
2)进行缩合反应得到希夫碱甲氧基甲基吲哚(
3),然后与炔丙基碘(
4)的格氏试剂反应生成炔丙基假吲哚(
5)。该物质在三氟化硼和氧化汞的催化下于甲醇中反应,经过甲基被迫处于顺式的中间体(
6)(亲电加成)发生关环反应得到(
7)。
D环合成
D环的合成是从手性的(S)-樟脑(
8)在羟胺作用下转化为肟(
9)开始的,然后通过水解转化为酰胺(
10),继续反应得到内酰胺(
11)(酸-胺缩合),N-亚硝基化合物(
12)、重氮化合物(
13)和环戊烯衍生物(
14)(卡宾甲基插入)。使用氢化铝锂还原得到醇(
15),再用铬酸氧化得到醛(
16),然后用(亚甲基甲酸甲酯)三苯基膦进行一次Wittig反应生成反式烯烃(
17),最后水解生成羧酸(
18)。
AD环偶联
胺(
7)和羧酸(
18)在酰氯作用下缩合成酰胺(
19),然后在叔丁醇中用叔丁醇钾处理,发生Michael加成反应得到氢原子处于反式的三环化合物(
20)。对将要进行芳香环部分还原的底物上保护基,其中一个羰基与乙二醇反应生成缩酮(
21)另一个与亚胺离子的盐(氟硼酸三乙基𨦡)在甲醇钠的甲醇溶液中反应生成烯醇醚(
22)和原酰胺(
23)。在甲苯中加热逸出甲醇可得到烯醇醚(
24)。Birch还原反应生成四烯(
25),再用酸处理得到二酮(
26),也被称为异环戊烯酮(
pentacyclenone)。
26中的第二个保护基乙缩醛通过酸解转换为酮(
27)。单肟(
28)(位阻比酮羰基更大)可以由二肟的选择性水解(亚硝酸或乙酸催化)制得。新的第二个氮原子是构建AD环关环时需要的。环戊烯环以及环己烯酮环经臭氧化反应氧化生成三酮(
29),然后1,5-二酮单元进行一次羟醛缩合反应(四氢吡咯乙酸酯)生成六元环(
30),并伴随着肟基的甲苯磺酰化,用高碘酸再次氧化打开六元环,最后用重氮甲烷酯化得到酯基(
31)。通过Beckmann重排(甲醇、聚苯乙烯磺酸钠、2小时、170 °C)得到未拆分的内酰胺(
32),它进一步经胺羰缩合反应、羟醛缩合反应得到四环化合物(
33)
α-咕啉去甲基甾酮(
α-corrnorsterone)(中文名称的来历详见注释)。这种内酰胺化合物不易开环,因为丙酸酯侧链的立体化学不相符。α化合物因此在过量的碱作用下被转化为它的差向异构体(
34),然后再次酸化并用重氮甲烷处理。差向异构体随后同时与甲醇和苯硫酚反应转化成
35。这确保了咪唑尾状长链的特异性。然后进行臭氧化反应生成醛(
36),同时氨将硫酯转化为酰胺,再用硼氢化钠进行醛的还原反应、甲磺酰化,然后用溴化锂溴化得到溴化物(
37)。最后一步将酰胺基脱水成腈基完成AD环合成。
C环合成
C环合成的起始原料是手性(+)-樟脑醌(
38),然后三氟化硼在乙酸酐存在时与之加成,使其转化为三甲基环己烯酸乙酯(
39),这个反应最早由玛拿西(
Manasse)和塞缪尔(
Samuel)于1902年首创。下一步酯水解成羧酸(
40),然后酰胺化成为酰胺(
41),再进行臭氧化反应得到有机臭氧化物(
42)。该物质可用锌和甲醇还原成丁二酰亚胺(
43),然后用盐酸的甲醇溶液处理生成内酰胺(
44),最后热解完成C环的合成(
45)。
B环合成
B环合成的起始原料是
3-甲基-4-氧代-2-戊烯酸(
46),它与丁二烯在氯化锡作用下经Diels-Alder反应形成外消旋的六元环化合物(
47)。该反应是立体专一性的,甲基和羧基处于顺式,此后这被证明是一个对旋电环化反应。使用α-苯乙胺进行手性拆分得到有光学活性的异构体(
47)。用铬酸氧化双键生成三元羧酸中间体(
48),经两次分子内酯化得到双内酯(
49)。通过一次Arndt–Eistert反应在羧酸α位插入一个亚甲基(
50),再与氨反应生成内酰胺(
51),最后与五硫化二磷反应获得硫内酰胺(
52)。
BC环偶联
B环(
52)和C环(
45)在过氧化苯甲酰和盐酸作用下形成硫桥键(
53)。先进行一次Eschenmoser去硫反应硫原子在亚磷酸三乙酯的位阻影响下形成烯胺和亚胺(
54),然后内酰胺基团在氟硼酸三乙基𨦡和硫化氢存在下转化为硫内酰胺(
55)。
AD、BC环偶联
图中右侧的BC分子(氰溴化物(
37))和左侧的AD部分(丙酸酯部分外消旋化的
硫代右前体(
55)(
thiodextrolin,名称的来历详见注释))在叔丁醇钾的催化下,经过一个硫离子中间体生成硫醚(
56)。然后使用氰乙基膦、三氟乙酸和环丁砜进行合成过程的第二次Eschenmoser去硫反应,得到
氰基咕啉内酯前体(
57)(
cyanocorrigenolide),同时C环的丙酸酯基团也被外消旋化。由于两部分巨大的空间位阻,这样的偶联是至今为止唯一成功的方法。内酰胺和内酯基团在五硫化二磷、4-甲基吡啶和氟硼酸三乙基𨦡作用下生成
二硫代氰基咕啉内酯前体(
58)(
dithiocyanocorrigenolide)和S-甲基衍生物(
59)。然后二甲胺与之加成并打开硫内酯环,并通过硫离子从甲基上的消除生成一个环外烯键。一个早期模板定向合成该化合物的例子以钴加合物的形式分离出它。
最后由(
60)到(
61)的成环反应是受形成钴配合物推动的,这是在二氮杂二环壬烷和二甲基甲酰胺的条件下进行另一种的去硫反应。该反应伴随着环C中丙酸酯基团的外消旋化。再用碘和乙酸氧化形成内酯(
62),并恢复B环的尾状丙酸酯基团的正确立体化学性质。
最后的努力目标是在5号位和15号位引入甲基。因为10号位被充分屏蔽,该物质与苄基氯甲基醚反应生成双(氯甲基)加合物,后者可用苯硫酚进一步转换成二硫代苯基化合物(
63),产物分离需要使用薄层色谱法。然后用雷尼镍加氢脱硫,该还原反应同时将内酯环打开使之成为羧酸,再用重氮甲烷与之反应生成酯(
64)。
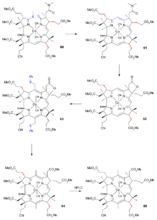 维生素B12AD、BC环偶联(二)
维生素B12AD、BC环偶联(二)
在这个阶段,混合物异构体的数目通过高效液相色谱法减少到两个,也就是(
65)中C环13号位的外消旋丙酸酯基团。与硫酸的反应将氰基转化为酰胺基(
66),但C环13号位的立体化学性质再次被破坏。产量较少的所需异构体(
67)再次用高效液相色谱法分离出来。酰胺基然后被来源于氯乙醛的环己基硝酮(
cyclohexylnitrone)和四氟硼酸银转化为羧基(
68)。最后一步中六个酯基与氨合氯化铵反应转化成钴啉胺酸中的酰胺基(
69)。
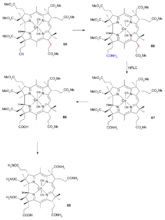 维生素B12AD、BC环偶联(三)
维生素B12AD、BC环偶联(三)
$firstVoiceSent
- 来自原声例句